

ここでは医薬品の開発プロセスについて解説します。
医薬品は様々な試験や課題をパスして初めて医薬品として承認されます。そのため、開発には多くの人材・職種が複雑に絡み合っています。
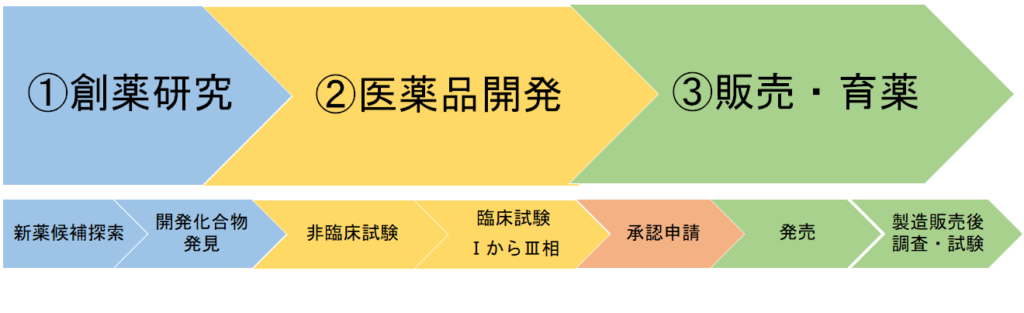
薬の一生を大きく分けると、①創薬研究、②医薬品開発、③販売・育薬となっています。
このカラムでは②医薬品開発と③販売・育薬の間、 承認申請 部分について解説します。
承認申請
承認申請 作業とは、入手した医薬品に関する様々な情報をまとめ、行政に情報を提出することです。
行政に審査をお願いするので、内容が纏まっていて確認しやすく、合理的な説明になっているとスムーズな審査が行われます。
・資料はシンプルに
・かつ合理的、妥当な説明
読みやすい文書であれば、スムーズな審査が行われます。
会社ごとの色が出る部分だといえます。
しかし、創薬作業のなかで”一番の山場”でである承認申請、なかなか一筋縄にはいかないのが普通です。
審査にかかる時間の標準目安として申請から約1年で承認となります。
途中、当局からの質問照会があり、中には想定していなかった指摘から、申請作業が長期化してしまうことも往々にしてありえます。
審査文書は専用フォーマットがあります。理由は形式の統一化による審査の迅速化が考えられます。
同じ形式なら審査側も内容確認がしやすいことは想像できますよね。
各企業の申請担当者は、専用の申請フォーマットに沿って情報を整理し、提出資料を作成します。
各項目の記載は抑えるべきポイントや、ある程度のルールがあります。
しかし、文章作成自体は自由であり、だからこそ十な合理的理解が得られる文書作成能力がカギとなります。
またもう一つ大きなポイントとして、承認申請とは、”ただの化合物”か”医薬品”かの転換点といえます。相応の管理が必要となる格上げの段階であり、前後での薬剤師としての関わり方も大きく変化します。
新薬の承認申請で必要な情報
製造販売承認申請書(以下、申請書)を作成し、提出しなければなりません。
申請書はそのまま、承認書に引き継がれる前提の内容であり、医薬品の製造レシピ、管理ブックの役割を果たします。
”本文”には
- どのように作られるか
- どのように分析して管理ができるか
- 管理者は誰か
等がメインに記載されています。具体的には
- 名称
- 成分及び分量又は本質
- 別紙規格
- 製造方法
- 用法用量及び効果
- 効能又は効果
- 貯蔵方法及び有効期間
- 規格及び試験方法
- 製造販売する品目の製造所
- 原薬の製造所
- 備考1,2
では、臨床試験の結果はどこに?と思われたと思います。
それらデータは添付資料として提出が義務付けられています。
承認後の維持管理として、申請データ自体はそれほど重要ではないため、本文ではなく”添付資料”としての位置づけとなっています。添付資料として必要な情報は下記の通り。
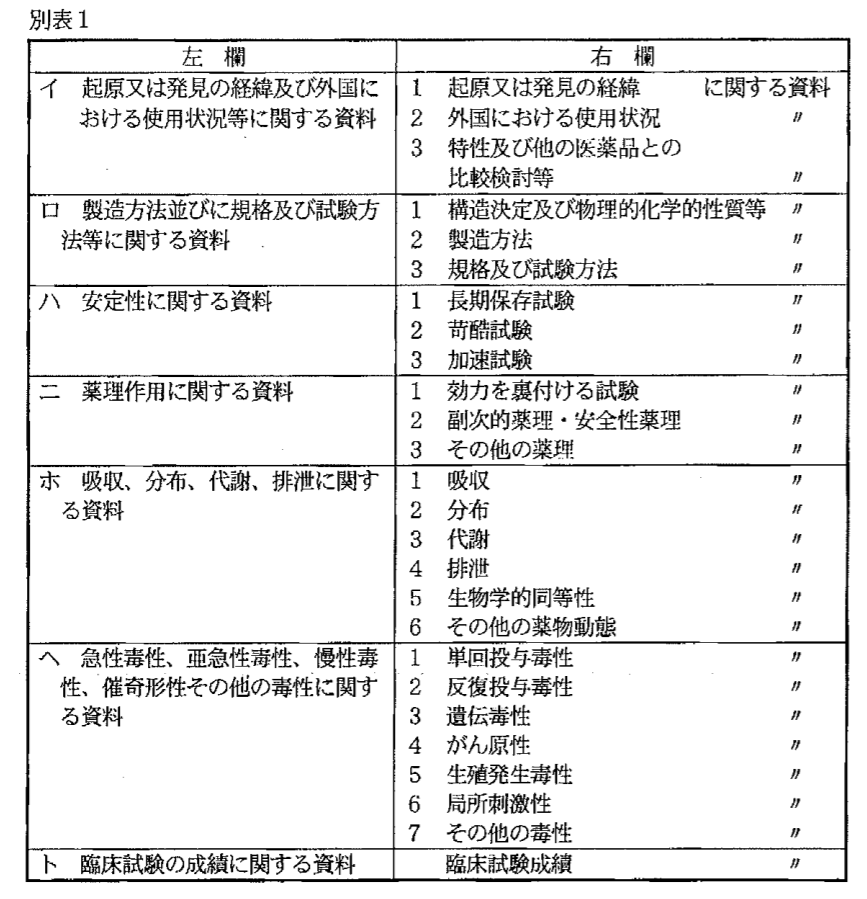
各情報はそれぞれが完結で合理的にまとめられたものが要求されます

承認申請の方法
医薬品等電子申請ソフト(FD申請書)
薬機法における申請書のフォーマットは薬機法施行規則に規定されています。
従来は手書きが主流でしたが、平成17年の旧薬事法改正に合わせて、フロッピーディスクやCDを使用した電子申請運用がスタートしています。
膨大な資料提出から、大幅に申請作業の負担が減ったことが想像されます。
申請以外の薬事文書作成に現在でも使用されているFD申請システムですが、設計がすでに16年前とかなり古いので、使用感といえば…なかなかの熟練度が必要なソフトとなっています。
一般用医薬品(OTC)は現在も同じソフトで資料作成となっています。
しかし、新規の医療用医薬品については、この申請方法ができません。
(数年前まで、医療用の後発品申請はFD申請が主流でしたが、現在はCTDへ移行しています)
CTD:コモン・テクニカル・ドキュメント(国際共通化資料)
Common Technical Documentの略称で、医薬品の承認申請のために作成する日米EU共通の国際共通化資料のフォーマットです。新薬はこちらを使用して申請します。
医薬品開発は医薬品の国際的な研究開発の促進、及び患者への迅速な供給のため、承認審査資料の国際的なハーモナイゼーション推進の必要性が指摘されていました。
そこで、日、米、 EU の医薬品規制調和国際会議(ICH)が組織され、新医薬品の承認申請資料の調和を図るための活動が行われています。
平成 12 年 11 月にCTDが合意されたことを皮切りに20年余りが経過している国際的な取り組みです。
共通化の大きな目的は
①フォーマットが統一され、時間・コスト軽減、内容確認しやすくなる。監査者視点も統一しやすい。
②膨大な治験データを電子的提出、管理ができる
③承認後のライフサイクルマネジメントに便利になった。(承認後の管理もラクになる)

CTDは大きく5部の構成となっています。
第1部(モジュール1)は資料全体の概略。申請する地域ごとに要求情報の特色が異なることが特徴。
第2部から第5部(モジュール2~5)は、全ての地域への申請において共通となるように意図されている。
第2部が申請内容の根幹となり、品質、非臨床、臨床試験の結果から医薬品として妥当である説明がなされます。3,4,5部はそれぞれの具体的なデータが収納されるイメージです。
かつては申請先の国ごとに、各国独自のお作法に合わせて用意する必要があり、審査に時間がかかっていました。このような理由から、有用な薬なのに、手続きが難航して使えない、いわゆる『ドラッグラグ』が大きな課題となっていました。
現在はルール発足から20年ほど経過し、ドラッグラグは大きく解消されている状態です。
しかしアジア諸国をも取りこんだ真の意味の統一は未だ達成されていません。
アジア諸国は経済的格差も大きく、社会水準もばらつきが多く標準化が難しい背景がありました。
ですが、徐々にASEANを中心としたアジア圏での共通方法の確立が進み、ACTDというASEAN共通の申請フォーマットも登場しています。
本家CTDとほぼ同じ構成ですが、多少アジア諸国の実情に合わせた部分もあります。
ICHを中心とした、本当の意味で審査レベルや審査スピードの共通化には、もう少し時間がかかるかもしれません。

薬剤師の活躍の場
申請書作成では、様々な部門からの情報を資料一つにまとめ上げる力が求められます。
特に全体総括の資料は、わかりやすい簡潔な記載が重要になってきます。
ライティングスキルの中には、日本語文章の作成能力はもちろん、
原料や試験方法の記載、公的規格書からの転用、日本薬局方記載総則など、資料特有の記載も学ぶ必要があります。
※ただし、そう難しいものではありません。
レギュラトリーサイエンスも知っておく必要があります。
意味は『科学技術の成果を人と社会に役立てることを目的に、根拠に基づく的確な予測、評価、判断を行い、科学技術の成果を人と社会との調和の上で最も望ましい姿に調整するための科学』だそう
少しわかりやすく説明すると、科学的知見や根拠と、規制などの行政施策・措置との間の橋渡しとなる科学のことです。
根拠が科学的に十分妥当であれば、それをベースに別ケースの良し悪しの判断材料になったり、確認作業が出来ますよね!?
薬事申請とはとても細やかな作業です。
あいまいな表現による操作ひとつが影響を及ぼすことも少なくありません。またそのような条件の結果を、他試験に当てはめることも比較困難になることが容易に想像できます。
つまり、妥当性ある範囲内の化学的知見から予測、評価、判断の蓄積をして他の科学技術の進歩にも生かす。という意味になります。
これら承認申請作業について、薬剤師の適性は高いと思っています。
経験がある程度必要な職種であり、薬剤師だけがこなせるというわけでもありません。しかし、多角的視点を持てる薬剤師にはやはり有利になる部分も多くあると思っています。
ここでは医薬品の開発プロセスについて解説します。 医薬品は様々な試験や課題をパスして初めて医薬品として承認されます。そのため、開発には多くの人材・職種が複雑に絡み合っています。 薬の一生を大きく分けると、①創薬研究、②医薬品開[…]

ここでは 医薬品開発 プロセスについて解説します。 医薬品は様々な試験や課題をパスして初めて医薬品として承認されます。そのため、 医薬品開発 には多くの人材・職種が複雑に絡み合っています。 薬の一生を大きく分けると、①創薬研究[…]

ここでは医薬品の開発プロセスについて解説します。 医薬品は様々な試験や課題をパスして初めて医薬品として承認されます。そのため、開発には多くの人材・職種が複雑に絡み合っています。 薬の一生を大きく分けると、①創薬研究、②医薬品開[…]


