

ここでは 医薬品開発 プロセスについて解説します。
医薬品は様々な試験や課題をパスして初めて医薬品として承認されます。そのため、 医薬品開発 には多くの人材・職種が複雑に絡み合っています。
薬の一生を大きく分けると、①創薬研究、②医薬品開発、③販売・育薬となっています。
このカラムでは②医薬品開発の部分について解説します。
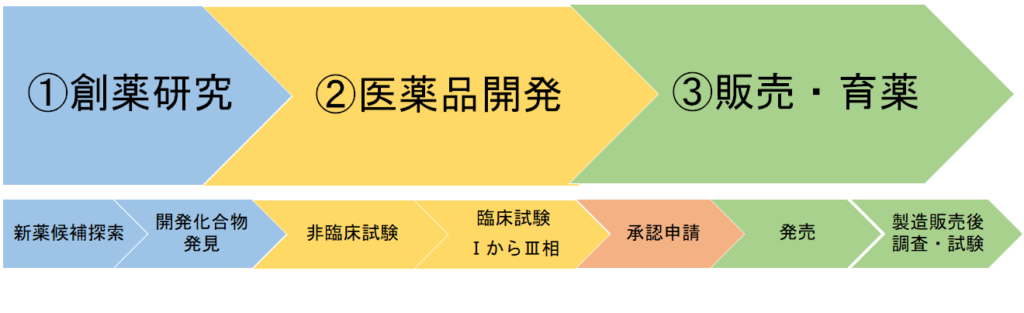
② 医薬品開発
シーズ化合物の中から、
医薬品を作ってみよう!と選ばれたものについて開発作業が開始されます。
薬として本当に効果があるか、安全なものかを検証する段階です。
後期においては、医薬品として安定供給するための製造方針検討も並行して行われます。
非臨床試験
非臨床試験とは、動物を使った投与実験の段階です。
候補物質の有効性・安全性・毒性などを調査する段階です。
下記の点について大まかに薬の特徴が明らかになっていきます。
- 薬力学(PD)、薬物動態学(PK)からADME:薬の効果と濃度の関係、薬を摂取してから排出するまでの流れ
- 薬効・薬理試験:培養細胞やモデル動物を使った効果測定
- 安全性試験(毒性試験):薬効を得る投与量よりも過剰な領域での安全性(毒性)の確認

対応する薬事関係法規
非臨床試験で得られたデータは、新薬申請時に提出するデータとなります。
そのためデータは信頼度が高いものでなければなりません。
試験は、確かな設備や確かな人員が揃っている施設で、妥当性がある然るべき方法、計画を元に実施されたものである必要があります。
そのような条件がそろった状態を「信頼性基準」と呼び、
特に安全性試験(毒性試験)には「GLP省令」が適用されます。
信頼性基準
医薬品申請に用いる薬理試験・薬物動態試験は信頼性の基準(薬機法施行規則第43条)に従い実施する必要があります。
主な要件は以下の通り。
・正確性:試験結果に基づき正確に作成されていること
・完全性、網羅性:有効性・安全性等を疑わせる結果が得られた場合でも、ありのままを記録すること。
・保存性:根拠となった資料が保存されていること
これらの観点が達成されているデータを用意しなければなりません。
GLP(Good Laboratory Practice)
非臨床試験において、
試験施設(場所)の設備・機器、組織・職員、検査・手順・結果等が、安全かつ適切であることを保証する基準です。
PMDAが施設調査し当該施設を認定しています。
GLPの基準をクリアする水準であることが認められると、GLP施設として認定を受けることができるのです。
GLPと「信頼性基準」の違いは??
日本においては
安全性試験にはGLP省令
薬理試験や薬物動態試験には信頼性基準
を適用することが定められています。
GLPは国際的なルールであるのに対し、信頼性基準は日本の薬機法で定められた独自のルールといえます。
しかし、
やるべきことのスタンスは実質同じなのですが、日本固有のルールが残っている状況です。
具体的な相違点は、GLPでは信頼性保証部門の設置や、当該部署による監査までが規定されています。
試験手技の実地確認等、信頼性保証部門の介入具合の手厚さの差を、GLP基準と信頼性基準として区別するケースが多数です。
しかし近年では、海外申請を検討する場合も多く、その際には実質GLP基準としての非臨床試験データが求められるケースも多いように思います。
それなら一見、GLPレベルの試験に統一するほうがメリットしかないように思えます。
しかし管理コストや人員の教育や確保などの運営コストが大きいため、GLP基準・信頼性基準といったグレード別体制が残っているように思えます。
製薬企業における職掌について
一般的に研究職と呼ばれるのは、非臨床試験段階までとされています。
以降の臨床試験は、ヒトを対象とした本格的な開発業務となるため、”開発職”と呼ばれます。
臨床試験
医薬品開発の上流では動物実験である程度の有効性、安全性が確認できた後、実際に人に対して使用するフェーズに進行します。
医薬品か初の後半である臨床試験は、ヒトを対象としてその有効性や安全性など、具体的な投与量から副作用まで、実用に向けたデータを取得、確認するために行われる試験となります。

治験
治験とは、国から医薬品や医療機器の製造・販売承認を得るために行う臨床試験のことです。
多くの場合、製薬会社が主体なっていますが、臨床研究に従事する医師が主導して実施される治験もあります。
※承認を得るための臨床試験を治験と呼び、市販後臨床試験は治験とは呼びません。
下記のように、徐々に検証目的を変えながら、対象者を増やしていく治験を実施していきます。

対応する薬事関係法規
治験をするにあたっては、非臨床と同様の実施に際する基準が設定されています。
GCPはその基準の一つであり、適切で妥当性がある試験を実施するための基準であるだけでなく、治験に参加する被験者の権利を最大限守るためのルールブックにもなっています。
GCP(Good Clinical Practice)
- 治験の内容を国に届け出る
製薬会社は、治験を担当する医師が合意した「治験実施計画書」を厚生労働省に届け出ます。
「治験計画書」には実際の服薬量や、回数、検査内容などが詳細に記載された文書です。この内容を厚労省が調査し、問題があれば変更等の指示を出します。 - 治験審査委員会(IRB)で治験の内容をあらかじめ審査すること
治験審査委員会は「治験計画書」について細かい設定を審査します。
・治験に参加する患者さんの人権と福祉を守っているか、
・試験が科学的に調査できる計画か、
・治験を行う医師は適切な人材か、
・参加する患者さんに治験内容を正しく説明するようになっているか
など、細かい設定を審査します。
また治験審査委員会には、医療専門外の者、病院と利害関係がない者が必ず参加します。
臨床試験とは様々な機関が絡み合います。
承認への道すがら協力してくれた施設への恩など、なにかとお金の話になりがちです。
治験審査委員会では、利益関係者だけの利潤を追求するような案件は排除し、医療倫理としての誠実性を重んじ被験者たちを守るための重要な仕組みとも言えます。 - 同意が得られた患者さんのみを治験に参加させる
症例数が欲しいがために勝手に治験に参加させてはなりません。 - 重大な副作用は国に報告する
お消えたことは包み隠さず開示する必要があります。 - 製薬会社は、治験が適正に行われていることを確認する
製薬会社の担当者(モニター)は、治験の進行を調査、「治験実施計画書」やGCPの規則を守って適正に行われていることを確認します。協力病院は全国各地に及ぶことも多く、出張が多くなる職といえます。
以上のように、患者の協力を得なければならないため、人権や個人情報など細心の注意が必要となる場面が多いのが臨床試験フェーズの特徴といえます。

臨床研究法
治験から少しズレますが、臨床研究法についても解説します。

臨床研究法
臨床研究とはヒトを対象とした試験であり、医薬品開発の研究手段としてとても重要です。
ただし臨床試験でデータがほしい企業と、実際の実施機関である医療機関の利害関係において、金銭的援助をもとに不正で恣意的な試験がなされる懸念がありました。
これを防止するための法律として臨床研究法が制定されています。
規制の対象となる主なポイントは以下の通り
- 製薬企業から資金提供があるか …提供を受けていてかつ、治験以外の臨床研究は特定臨床研究として対象
- 治験と同等の「実施基準」順守を義務化しているかどうか
- 実施計画の届出を義務化し 「認定委員会」で審査
- 研究の改善・停止を厚労省が命令
- 資金提供の公表 製薬企業に義務付け …不正な利潤などが無いよう、透明性の確保
今般、医薬品等を人に対して用いることにより、その医\薬品等の有効性・安全性を明らかにする臨床研究を法律の対象とすることとし、臨床研究の対象者をはじめとする国民の臨床研究に対する信頼の確保を図ることを通じてその実施を推進し、もって保健衛生の向上に寄与することを目的として、臨床研究の実施の手続、認定臨床研究審査委員会による審査意見業務の適切な実施のための措置、臨床研究に関する資金等の提供に関する情報の公表の制度等を定める「臨床研究法」が平成29年4月14日に公布され、平成30年4月1日に施行されました。
厚生労働省HPより
医薬品の承認申請に必要な試験はGCPに基づき的確に試験が実施されていましたが、
付加価値創出のためなど承認申請の本筋と少しズレる臨床検査においては、うやむやだったのが現状でした。
先のARB「ディオバン」をめぐる臨床研究データ改ざん事件では、そのような”直接に申請に使用しない枝葉の試験”にて自社に有利となる恣意的結果の選択が明らかとなったため、周辺関連試験も治験と同等の管理基準をもって実施することを義務付けたた法律となりました。
治験薬の製造について(治験薬GMP)
GCP省令に基づく治験に使用する薬は、まだ承認されていない薬とはいえ、適当な品質者を服用させるわけには行きません。
そのため治験薬GMPという特殊なものづくり基準の適用をもって、一定の製造水準を満たすことが必要となります。
そもそも臨床試験の時点では、剤型などが複数検討されるなど、決定しきれていない場合があります。
ですが治験に使用するにあたって、治験薬の品質にばらつきがあると、治験結果の妥当性に疑義が入るなどの懸念があります。
また、ゆくゆく市販を想定するならば、なるべく市販品と同形態として治験を行ったほうが、効果検証の意義、その後の管理や適合拡大の育薬作業にもつながり大変有益です。
治験薬の場合、未だ医薬品として成り立っていない段階でありますが、一定の品質水準を満たした製剤の準備が必要なため検討すべき要素となっています。
承認申請と市販後の製造を見越して
治験薬を一定の水準で製造する必要があるうえで、承認申請を見据えた製造方法の検討もスタートし始めます。
なぜなら、承認申請時に必要な内容は
①有効成分自体の効果、安全性
②実際の作り方、その評価の仕方
以上の2つが必ず必要になっています。(後発品の場合は②だけになります)
加えて審査完了後、申請書通りの製造が可能か、実地の製造確認も予定されています。
つまり、申請時点で製造のことまで何もかも決めておかなければならないのです。
試験管レベルで製造していた有効成分を工業用にスケールアップが必要であったり、より適切な剤型工夫となる製剤研究、製剤の評価分析法として適切な試験系を検討するなど、様々な準備があります。
医薬品開発 の分野で薬剤師は活躍できるか。
医薬品開発分野は、大規模な試験も増えるため関連する人数も大幅に増えていきます。
具体的な職種について解説します。
非臨床試験
非臨床試験は動物実験が主体です。薬効の評価には病態モデル動物の作成が欠かせません。
病態モデルとは疑似的に疾病症状を誘発させた動物の作成です。
例えば、癌の薬を投与するために疑似的に癌を発生させり、うつ病に対する薬の開発に際しては、動物にうつ症状を誘発させるような仕組みを検討します。
投薬後の評価として動物から採血、病変組織の病理解剖が必要だったり、結論としては生物学、病理学専攻者や獣医師の活躍の場が多いように感じます。
薬物動態などの評価においては分析研究者が多数活躍しています。
低分子化合物に代表される測定法はHPLCやMSを使用したクロマトグラフィーが汎用されています。高分子、中分子医薬品となると、抗原抗体反応を使用したイムノアッセイ系などの測定分析法も多数行われています。
機器分析において、薬剤師であればそのは下地があり、多角的視点で化合物を評価できる人材として活躍の場の1つとなっているように思います。
また、最も重要な視点として、薬事法施工令であるGLP省令をしっかり理解する必要があります。薬機法の下地が法の意図の理解、計画立案が可能になるため、薬剤師にはアドバンテージがある領域だと思います。
臨床試験
臨床試験は協力施設も一気に増えるため、さらに多くの方が関わる領域です。
臨床開発職
臨床試験において主役は臨床開発職です。主に製薬企業が務めます。一言でまとまっていますが、その職掌は多岐にわたります。
- 開発企画
- PM(プロマネ):全体の進行管理
- CRA(臨床開発モニター):医療機関も窓口となり、治験の進行状況をモニタリング。症例報告の書確認等。
- DM(データマネージメント):症例報告書の確認、解析用にデータ化
- BS(統計解析):DM作成のデータをもとに統計評価
- メディカルライティング:試験結果をまとめ、承認申請思資料を作成する。
- QC(Quality Control:品質管理):実施している治験に違反が無いか確認管理
- QA(Quality Assurance:監査):第三者視点として、実施された治験が問題なかったどうかを確認。
- 開発薬事:治験前相談、承認申請書類を提出、照会対応などの当局対応
- PMS(Post Marketing Surveillance):販売が開始された医薬品の有効性・安全性の確認と、市販前の治験で得られなかった新たな作用・副作用に関する情報収集のために行われる調査の総称。GPSP,GVPに規定。
治験をやって、結果の収集解析、まとめ作業と…言うは易しですが、様々な職掌に分担されています。
それほどに緻密な作業が要求されるということになります。しかし、創薬業界もアウトソージングが進み、自社で全てを実施する製薬企業は現在はほぼ皆無かもしれません。
医療従事者との面談や、データの取り扱いなど多岐にわたります。GCP省令に基づいた作業としては、薬剤師としてのバックグラウンドは非常に役立つといえるでしょう。
※医薬品承認を得るとは、多岐に渡る要件が必要であることから職種が多彩なのに対し、広く”臨床開発職”と1つのワードで規定されることも多く、不明瞭な情報も多いのが特徴です。別の記事で承認作業にフォーカスした職種の記事もあるので、併せて読んでもらえると幸いです。
CRO (Contract Research Organization):医薬品開発業務受託機関
CROとは製薬企業の開発業務を受託実施する機関です。
製薬企業の中にはほとんどのの業務をCROに業務委託をしているケースも増えてきました。
理由として、医薬品開発のコストが外注だと抑えられることや、CRO業界の成熟によ、品質も高いものが得られるようになっているからです。
CROに勤めるメリットは、多数の会社からの業務委託をしてるので、多岐に渡るノウハウや経験が得られるチャンスがあるといえます。
CRC(Clinical Research Coordinator):治験コーディネーター
CRCとは、治験を行う際に医療機関と製薬会社と患者の間に立ち、全体進行がスムーズになるようサポートする職種です。
SMO(Site Management Organization):治験施設支援機関に所属して医療機関へ派遣されるパターンと、大学病院などの医療機関に所属して院内の治験コーディネーターとして業務を行うパターンがあります。
医療従事者、メーカーの担当者、患者という三様のコミュニケーションや知識が必要な職種であり、三者の立場を理解できる薬剤師のうってつけの活躍の場とも言えます。
製造部門
製造部門の主たる活躍は承認後かもしれませんが、承認前も決して暇なわけではありません。
製剤研究、製剤技術
医薬品は様々な形態のものが存在します。
なんだか単純に、有効成分だけを摂取すればいいと思われますが簡単ではありません。内服で摂取が不可なら注射が必要だし、注射といっても注入した傍から効果を持たせたいのか否か様々です。
医薬品はそれぞれ最適な形で、最適な場所へ届けて効果を発揮することが理想です。
それらを考慮しているのが製剤研究だといえます。
例えば同じ成分でも、製剤技術の進歩で飛躍的に効果が上がったり、服用頻度を抑えられるなどの価値を生み出すことさえあるのです。
そんな製剤技術も同時に進行しているのです。
製剤技術は薬学生に活躍のメリットが大です。一番の強みは領域アドバンテージが高いこと。
薬を運ぶ形態の研究として近い分野専攻になるのは、薬学部以外には工学部(モノづくり系)しかないためです。
しかも生体に服用した場合の生物学的な要素、ミクロな世界の化学的素養を両立できる分野は薬学部を置いて他にないといってもよいかもしれまん。それほどまでに重要な職種といえます。
※薬の構成については別の記事でもまとめています。
生産管理、生産技術
臨床試験までの治験薬の製造量は実はたかが知れています。
ですが承認後には日本中、極論世界中の人に供給する可能性が発生するとかんがえれば、より大きく安定的に医薬品を作る方法が必要です。
では単純に試験管クラスから、成分比だけを変えずに大きくすることなんて、そんなの超然簡単…では全くないのです。
身近な経験から例えば、ゆで卵の作り方として”6分沸騰したお湯で煮る”が僕自身最適な条件だと思っていました。しかし…
- 同時に何個作るか
- どこで作るか(使用するコンロ、熱源のちがい)
- どんな方法で作るか(水から煮るのか、熱湯に投下するのか)
- どんな鍋で作るか
等をかんがえると実は条件はとっても多くて複雑。
実際に自宅ではベストタイミングでも、別の場所では全くうまくいかないことが沢山ありました。
医薬品も同様で比率だけの条件で同じものを作るのは困難です。
機器の能力や反応の条件等を加味しながら調整し、それでもうまくいかない場合は反応の主たるキーポイントを理解しないと解決ができない場合もあります。
薬剤師も適性としてアドバンテージがある職種といえます。
化学素養はもちろん、総量数トンを超す製造も珍しくないため物理的素養も必要になってきます。
加えて特に近年では抗体医薬など生物学的手法を用いた製剤も多数あります。様々な条件を多角的に観察できる点こそ、薬剤師のアドバンテージといえます。
分析、CMC薬事
医薬品は作りっぱなしではダメで、『我こそは”A”という医薬品である』という特徴、性格を明らかにする必要があります。そこに必要なものが分析技術です。
分析は言われた通りの方法で分析してるだけではないのです。
日本薬局方というルールブックにおける試験法に乗っ取った分析はもちろんですが、当然その方法では物性をとらえきれない化合物も存在します。
その化合物だと同定が可能である”確認試験”から、どの程度含まれているか測定する”定量法”に至るまでの方法をすべて設定しなければなりません。つまり分析とは化合物のアイデンティティを捉えるための適切な方法の確立こそが最大の役目になります。
CMC薬事は(Chemistry, Manufacturing and Control)を意味しさらに領域を広げます。
分析とは化合物、あるいは製品の特長を捉えることですが、分析方法の確立を含め、製品全体として安定的に生産が可能であること踏まえた管理を目的としています。
たとえば分析方法がとても優秀だったとしても、原料や製造環境などによる製造ロットごとのバラつきが発生することも少なくありません。こうした現象は有効性や安全性に対する評価に影響を与える可能性があります。
製造物の処方や規格およびそれらの評価方法や設定根拠から、使用する包材、原材料の管理、製造プロセスを検討し、バラつきを踏まえた上での製造物の品質評価を統合して行う概念となっています。
製品の製造揺らぎも併せて全体像として製造品質・条件を考える職種といえます。
多くの場合、企業における求人では、上記概念を踏襲したうえで、管理ハンドリングのし易い承認書の記載設定、その折衝作業を含んでいます。
薬剤師の適性については、最上レベルのアドバンテージがあるといえます。
分析は基本的素養があることが重要で、技術自体はマニアックな世界です。
また、CMC薬事となると、医薬品製造を俯瞰で見る素養が必要であり、メディカルライティングのスキルから、行政折衝の能力まで幅広く活躍のチャンスがあるといえます。


